Therapeutic potential of targeting kinase inhibition in patients with idiopathic pulmonary fibrosis
Article information

Abstract
Fibrosis is characterized by excessive accumulation of extracellular matrix components. The fibrotic process ultimately leads to organ dysfunction and failure in chronic inflammatory and metabolic diseases such as pulmonary fibrosis, advanced kidney disease, and liver cirrhosis. Idiopathic pulmonary fibrosis (IPF) is a common form of progressive and chronic interstitial lung disease of unknown etiology. Pathophysiologically, the parenchyma of the lung alveoli, interstitium, and capillary endothelium becomes scarred and stiff, which makes breathing difficult because the lungs have to work harder to transfer oxygen and carbon dioxide between the alveolar space and bloodstream. The transforming growth factor beta (TGF-β) signaling pathway plays an important role in the pathogenesis of pulmonary fibrosis and scarring of the lung tissue. Recent clinical trials focused on the development of pharmacological agents that either directly or indirectly target kinases for the treatment of IPF. Therefore, to develop therapeutic targets for pulmonary fibrosis, it is essential to understand the key factors involved in the pathogenesis of pulmonary fibrosis and the underlying signaling pathway. The objective of this review is to discuss the role of kinase signaling cascades in the regulation of either TGF-β-dependent or other signaling pathways, including Rho-associated coiled-coil kinase, c-jun N-terminal kinase, extracellular signal-regulated kinase 5, and p90 ribosomal S6 kinase pathways, and potential therapeutic targets in IPF.
Introduction
Fibrosis is an excessive deposition of extracellular matrix (ECM) components, particularly fibrillar type I and III collagen [1,2]. Fibrosis is mainly driven by profibrogenic and proinflammatory cytokines, including the transforming growth factor beta (TGF-β) superfamily, tumor necrosis factor alpha (TNF-α), various interleukins, oxidative stress, and inflammation [3,4]. As a result, fibrosis can lead to failure of vital organs, including the lung, liver, heart, kidney, skin, and eye [5]. Pulmonary fibrosis is the final outcome of various parenchymal lung disorders, known as interstitial lung disease (ILD) [6]. One of the most common subtypes of ILD is idiopathic pulmonary fibrosis (IPF), which is a chronic, progressive, and generally fatal parenchymal lung disorder of unknown cause, with an approximate median survival of 2 to 5 years from diagnosis [7]. The clinical characteristics of IPF are heterogeneous and unpredictable, mainly including chronic cough, exertional dyspnea, declining lung function, and poor quality of life [8]. Epidemiological studies indicate that IPF is an age-related disease, and the majority of cases are diagnosed in patients over 60 years of age [9].
The pathogenic mechanism in IPF is not clearly defined, but the disease is characterized by epithelial injury and activation, epithelial-mesenchymal transition (EMT), sustained fibroblast activation, and excessive ECM accumulation, which result in progressive and irrevocable distortion of the histological lung structure [10,11]. Previous studies have revealed the complex and vital role of TGF-β/Smad signaling in lung fibrosis [12-15]. Enhanced TGF-β1 signaling with excessive ECM accumulation has been reported in experimental models of pulmonary fibrosis as well as in human lung fibrotic tissue [16,17]. The inhibition of TGF-β by neutralizing anti-TGF-β antibody, decorin, RNA interference, or antisense oligonucleotides alleviates fibrosis [18-21]. Moreover, in a mouse model of bleomycin-induced pulmonary fibrosis, Smad3 deficiency attenuated pulmonary fibrosis [22]. TGF-β signaling can initiate both canonical Smad-dependent and Smad-independent signaling pathways [23]. In Smad-independent pathways, TGF-β activates the phosphoinositide 3-kinase (PI3K)/Akt pathway and mitogen-activated protein kinases (MAPKs) such as extracellular signal-regulated kinase (ERK) 1/2, p38, and c-jun N-terminal kinase (JNK) 1/2/3 [24].
It has long been assumed that acute and chronic alveolitis lead to a fibrogenic response and play a critical role in the disease progression of IPF [25]. There are two different mechanisms involved in the pathogenesis of IPF. One of these is the ‘inflammatory pathway,’ which represents the major etiological pathway for IPF, associated with a marked collapse in the integrity of alveolar epithelial cells and subsequent fibrotic stage [25]. The other is the ‘epithelial/fibroblastic pathway,’ revealed by IPF [26]. These pathological changes, along with the disruption of the epithelial basement membrane enhance the migration of fibroblasts/myofibroblasts into the alveolar spaces and their subsequent deposition into the intra-alveolar ECM [26,27]. Many studies have shown that injured/activated alveolar epithelial cells in lungs from patients with IPF produce a variety of growth factors and pro-fibrotic cytokines [27,28].
Clinical treatment for pulmonary fibrosis
Pirfenidone and nintedanib have been recently approved for the treatment of IPF [29-31]. Pirfenidone is a small molecule that inhibits inflammatory responses and the progression of fibrosis in experimental models and patients with IPF [32-34]. It downregulates the proliferation of fibroblasts and TGF-β1-induced collagen synthesis and reduces the production of the inflammatory cytokine TNF-α and interleukin-1β both in vitro and in vivo [35]. In a phase 3 study comparing pirfenidone with placebo in patients with IPF, pirfenidone treatment for 52 weeks significantly prolonged progression-free survival, compared with placebo [36]. In addition, with pirfenidone, there was a relative reduction of 47.9% in the proportion of patients who had a decline in predicted forced vital capacity or who died [36]. Pirfenidone is frequently associated with gastrointestinal adverse effects such as dyspepsia, nausea, and gastritis [37].
Nintedanib is a small-molecule tyrosine kinase inhibitor targeting fibroblast growth factor receptor (FGFR) 1–3, vascular endothelial growth factor receptor (VEGFR) 1–3, and platelet-derived growth factor receptor (PDGFR) αβ, which are potentially involved in the progression of pulmonary fibrosis [38]. Nintedanib inhibits FGFR and PDGFR autophosphorylation and subsequent activation of downstream signaling via the Ras/Raf/MAPK, ERK1/2, and PI3K/Akt pathways [38]. Vascular endothelial growth factor (VEGF) stimulates angiogenesis through VEGFR and also binds to PDGFR in fibroblasts, subsequently stimulating cellular proliferation [38]. Nintedanib reduces migration, proliferation, and survival of fibroblasts, and ultimately attenuates angiogenesis in the lung [39]. In addition, administration of nintedanib attenuated the histopathological features of pulmonary fibrosis and expression of profibrogenic genes in experimental models of lung fibrosis [40]. In the two replicate phase 3 trials, nintedanib was shown to slow disease progression in patients with IPF by decreasing the annual rate of decline in forced vital capacity [41].
Other recommendations for the pharmacological treatment of pulmonary fibrosis are warfarin, N-acetyl cysteine, imatinib, and endothelin receptor antagonists [9]. However, most clinical trials did not show significant differences between placebo and treatment effects in patients with IPF [9]. Therefore, there is still a need to develop new therapeutic targets and agents to inhibit the progression of pulmonary fibrosis and improve mortality rates.
Clinical trials with kinase inhibitors for idiopathic pulmonary fibrosis
1. Receptor kinases
In recent years, growth factors and receptor kinases have attracted attention as potential drug targets for pulmonary fibrosis. Several therapies targeting receptor kinases, including growth factor receptors, are currently in clinical trials for IPF (Table 1). Aberrantly activated lung epithelial cells are the primary source of TGF-β, fibroblast growth factor (FGF)-2, PDGF, connective tissue growth factor (CTGF), and endothelin-1, key factors in the development of IPF. Based on this evidence, clinical trials of nintedanib targeting multiple growth factor receptors could be successful. Additionally, several clinical trials of cytokine receptors, including TNF-α, interferon-γ, and interleukin-13, have been conducted with no significant impact in patients with IPF (NCT 02277145, NCT00532233, and NCT00075998).

Featured clinical trials targeting kinases in patients with idiopathic pulmonary fibrosis
TGF-β mediates tissue fibrosis via recruitment and activation of monocytes and fibroblasts, and production of ECM through activation of serine/threonine kinase receptors [42,43]. In addition, TGF-β1 regulates the proliferation, differentiation, apoptosis, adhesion and migration, immunity, and even embryonic development, which ultimately contribute to fibrogenesis [44]. TGF-β has been shown to drive fibroblast-to-myofibroblast differentiation and directly promote pulmonary fibrosis in a mouse model of IPF [45]. Although TGF-β1 causes tissue fibrosis mainly by stimulating its downstream Smad signal transduction pathway, it is also known to activate Smad-independent signaling pathways, including MAPKs, focal adhesion kinase, and PI3K-Akt cascades in the pathogenesis of pulmonary fibrosis [46-48]. Both pharmacological and genetic inhibition of PI3K reduced pulmonary fibrosis in experimental rodent models, whereas overexpression of PI3K was observed in lung tissues from patients with IPF. A phase 1 clinical trial with a pharmacological inhibitor of PI3K is being conducted in healthy male and female subjects (NCT03502902).
Drug development has been challenged by the problem of identifying selective pharmacological inhibitors of the TGF-β1 signaling pathway that function by inactivating either the ligand or receptor of TGF-β1. Since TGF-β family members are secreted in the form of inactive complexes with latency-associated peptide (LAP), which binds to integrin αVβ6, inhibition of the binding between αVβ6 and the LAP region of TGF-β1 has been considered as a potential strategy for drug development in IPF [49]. A couple of phase 2 clinical trials of an immunoglobulin G monoclonal antibody and a small-molecule inhibitor of integrin αVβ6 are being conducted in patients with IPF (NCT01371305 and NCT04396756). In addition, an inhalation formulation of a nucleic acid medicine that selectively suppresses the expression of TGF-β1 has been tested in a phase 1 clinical trial in patients with IPF (NCT03727802). In contrast to those of TGF-β ligand inhibition, there are no active clinical trials of direct inhibitors of TGF-β1 receptor in patients with IPF. There is, however, an ongoing phase 2 clinical trial of a galectin-3 inhibitor that indirectly suppresses TGF-β signaling via reduced cell surface expression of TGF-β receptors (NCT03832946) [50].
CTGF, also known as cellular communication network factor 2, is a multifunctional growth factor that has been implicated in cell migration, proliferation, differentiation, and angiogenesis [51-54]. Since CTGF is an immediate early gene induced by TGF-β, PDGF, FGF-2, VEGF, and hypoxia, CTGF could regulate ECM deposition, tissue remodeling, and neovascularization, leading to the development of tissue fibrosis [55-58]. CTGF binds to integrin receptor α5β1 and induces the transactivation of FGFR2, PDGFR, and TGF-β receptor [59]. The fact that CTGF was elevated in lung fibrosis model and also in patients with IPF suggests its potential role in the treatment of IPF [60]. The neutralizing monoclonal antibody for CTGF has been shown to reduce lung fibrosis in experimental models [61,62]. A phase 3 clinical trial of a monoclonal antibody for CTGF is progressing in patients with IPF (NCT03955146).
2. Intracellular kinases
Intracellular kinases are attractive targets for the treatment of IPF. There are a couple of clinical trials of inhibitors of the Rho-associated coiled-coil kinase (ROCK) and JNK (Table 1). The regulation of the actin cytoskeleton is a major feature of chronic fibrotic diseases implicating the wound healing process against tissue injury [63,64]. The ROCK family of serine/threonine kinases are key regulators of profibrotic processes and reasonable targets for a new therapeutic strategy for pulmonary fibrosis [65]. ROCK activation has been observed both in patients with IPF and in a mouse model of lung fibrosis, and pharmacological inhibition of ROCK protected mice from experimental lung fibrosis [66]. A phase 2 clinical trial of a pharmacological inhibitor of ROCK is ongoing in patients with IPF (NCT02688647) [67].
It has been suggested that JNK activation in multiple cell types involved in lung fibrosis is positively correlated with the degree of fibrosis [68]. JNK1-deficient mice showed improved lung function in experimental models of lung fibrosis [69,70]. In a house dust mite model of lung fibrosis, a pharmacological JNK inhibitor decreased ECM accumulation and fibrosis [71,72]. A phase 2 clinical trial with a small-molecule inhibitor of JNK is being conducted in patients with IPF (NCT03142191).
ERK5 and p90 ribosomal S6 kinase (p90RSK) in the fibrotic response
A multitude of profibrotic mediators, including TGF-β, CTGF, PDGF, and FGF, and their signaling cascades, play an important role in the pathogenesis of fibrotic lung diseases. Collectively these signs of progress imply that kinase can be a good therapeutic target for pulmonary fibrosis. It has been shown that MAPK kinase (MEK) 1/2-ERK1/2-p90RSK inhibition reduces PDGF-AA-induced cellular migration [73]. FGF-2, a potent mitogen for fibroblasts, induces the synthesis of collagen in lung fibroblasts and myofibroblasts. Inhibition of ERK1/2 suppresses FGF-induced DNA synthesis, phosphorylation of ERK1/2, and p90RSK [74]. VEGF also causes rapid activation of Raf-1, MAPK, p90RSK in cardiac myocytes, and fibroblasts [75]. In addition, ERK5 modulates PDGF-induced proliferation and migration of hepatic stellate cells [76]. Many studies have revealed that ERK5 activation is induced by growth factors such as epidermal growth factor (EGF), FGF-2, and VEGF [77]. Thus, it is interesting that ERK5 is a common combined target for the treatment of pulmonary fibrosis through the regulation of growth factor signaling (Fig. 1A).
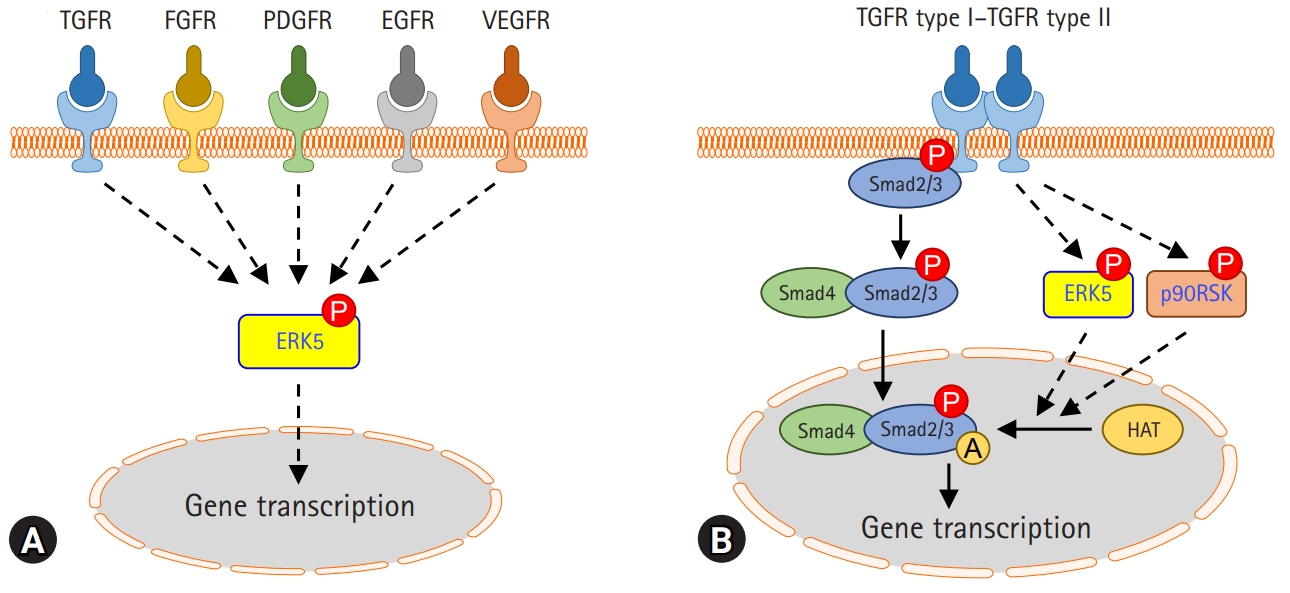
Roles of extracellular signal-regulated kinase 5 (ERK5) and p90 ribosomal S6 kinase (p90RSK) in pulmonary fibrosis. (A) ERK5 may be activated by multiple receptors involved in pulmonary fibrosis. (B) Transforming growth factor beta (TGF-β) activates ERK5 and p90RSK in lung alveolar epithelial cells and lung fibroblasts. Activation of ERK5 or p90RSK regulates Smad3 transcriptional activity via acetylation modification. The pharmacological inhibitors of ERK5 or p90RSK reduce TGF-β-induced fibrogenic gene expression and experimental lung fibrosis. TGFR, transforming growth factor receptor; FGFR, fibroblast growth factor receptor; PDGFR, platelet-derived growth factor receptor; EGFR, epidermal growth factor receptor; VEGFR, vascular endothelial growth factor receptor; P, phosphorylation; A, acetylation; HAT, histone acetyltransferase.
ERK5 is an atypical member of the MAPK family and plays a critical role in hypertrophic cardiac remodeling via regulating fibrotic genes and ECM expression [78]. ERK5 is also involved in the enhancement of cell viability and ECM accumulation in chronic glomerulonephritis [79]. Since ERK5 could be activated by various growth factors affecting pulmonary fibrosis, it has been investigated whether ERK5 regulates TGF-β1-induced profibrotic responses and the pathogenesis of pulmonary fibrosis. Kim et al. [80] reported that pharmacological inhibition of MEK5/ERK5 with BIX02189 and depletion of ERK5 using small interfering RNA against ERK5 inhibited TGF-β1-induced ECM production and Smad3 transcriptional activity, but not Smad3 phosphorylation and nuclear translocation. Notably, it has been shown that ERK5 plays a vital role in TGF-β1-induced fibrogenic signaling via enhancing Smad3 acetylation [80]. Moreover, the pharmacological inhibition of ERK5 ameliorated lung fibrosis and improved survival rate in a mouse model of bleomycin-induced lung fibrosis [80]. This suggests that ERK5 may provide a potential therapeutic strategy to prevent the progression of pulmonary fibrosis (Fig. 1B).
p90RSK is a family of serine/threonine kinases that is activated by the extracellular signal-regulated kinase signaling pathway. p90RSK is involved in numerous signal transduction and regulation of diverse cellular processes, including cell proliferation, growth, apoptosis, and transformation [81]. A recent study proposed that p90RSK is involved in the development and progression of liver fibrosis and hepatocellular injury in chronically damaged livers [82]. In addition, it has been reported that pharmacological inhibition of p90RSK using kaempferol inhibits TGF-β1-induced EMT and migration of A549 lung cancer cells [83]. A recent report showed that pharmacological inhibition of p90RSK by fluoromethyl ketone (FMK) or genetic inhibition of p90RSK significantly inhibited TGF-β1-induced Smad3 transcriptional activity, but not Smad3 phosphorylation and nuclear translocation [84]. In an experimental mouse model of bleomycin-induced lung fibrosis, p90RSK inhibitor FMK reduced pulmonary fibrosis, which suggests that it may be a novel therapeutic target for the treatment of lung fibrosis (Fig. 1B).
Conclusion
Pulmonary fibrosis is a dreadful condition that demands urgent attention. Although TGF-β1 is known to play a critical role in the pathogenesis of pulmonary fibrosis, clinical trials of therapies targeting TGF-β are progressing through repeated failures. According to recent data, kinase inhibitors have been identified as reliable targets for developing therapeutic drugs to treat IPF through regulation of not only TGF-β signaling but also multiple kinase cascades. A couple of receptor kinases are progressing for clinical trials in patients with IPF. In addition to clinical trials, recent preclinical studies with an experimental mouse model of bleomycin-induced lung fibrosis in our group have suggested that pharmacological inhibition of ERK5 or p90RSK could be a potential target of pharmacological treatment of pulmonary fibrosis through inhibition of TGF-β-induced Smad3 transcriptional activation. Further intensive studies using selective kinase inhibitors are needed to develop therapeutic agents that might slow the progression of the disease and improve the prognosis of IPF.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study was supported by the Basic Science Research Program (NRF-2018R1A2B6004664 to CHW, NRF-2018R1D1A1B07048399 to JHL) through the Korean National Research Foundation (NRF) funded by the Ministry of Science, ICT, and Future Planning, and by the Basic Science Research Program through the NRF funded by the Ministry of Education (NRF-2019R1I1A1A01060129 to SK).
Author contributions
Conceptualization, Investigation, Funding acquisition: all authors; Writing-original draft: SK; Writing-review & editing: JHL, CHW.