E-Submission
E-SubmissionPubMed Central, CAS, DOAJ, KCI

Articles
- Page Path
- HOME > J Yeungnam Med Sci > Volume 39(2); 2022 > Article
-
Case report
Idiopathic multicentric Castleman disease presenting progressive reticular honeycomb infiltration of lung and immunoglobulin G and immunoglobulin G4 dominant hypergammaglobulinemia: a case report -
Hyun-Je Kim1
 , Young-Hoon Hong2
, Young-Hoon Hong2 -
Journal of Yeungnam Medical Science 2022;39(2):153-160.
DOI: https://doi.org/10.12701/yujm.2021.01039
Published online: July 5, 2021
1Division of Rheumatology, Department of Internal Medicine, CHA Gumi Medical Center, CHA University, Gumi, Korea
2Division of Rheumatology, Department of Internal Medicine, Yeungnam University College of Medicine, Daegu, Korea
- Corresponding author: Young-Hoon Hong, MD, PhD Division of Rheumatology, Department of Internal Medicine, Yeungnam University College of Medicine, 170 Hyeonchung-ro, Nam-gu, Daegu 42415, Korea Tel: +82-53-620-3841 Fax: +82-53-654-8386 E-mail: yhhong@med.yu.ac.kr
Copyright © 2022 Yeungnam University College of Medicine, Yeungnam University Institute of Medical Science
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 4,965 Views
- 90 Download
Abstract
- Multicentric Castleman disease (MCD) is an uncommon systemic lymphoproliferative disorder that may cause multiple organ damage. Castleman disease-associated diffuse parenchymal lung disease (DPLD) has not been well studied. A 32-year-old man was referred to our hospital for progressive generalized weakness, light-headedness, and dyspnea on exertion for more than one year. Laboratory evaluations showed profound anemia, an elevated erythrocyte sedimentation rate, and an increased C-reactive protein level with polyclonal hypergammaglobulinemia. Chest radiography, computed tomography (CT), and positron emission tomography-CT scan demonstrated diffuse lung infiltration with multiple cystic lesions and multiple lymphadenopathy. In addition to these clinical laboratory findings, bone marrow, lung, and lymph node biopsies confirmed the diagnosis of idiopathic MCD (iMCD). Siltuximab, an interleukin-6 inhibitor, and glucocorticoid therapy were initiated. The patient has been tolerating the treatment well and had no disease progression or any complications in 4 years. Herein, we report this case of human herpesvirus-8-negative iMCD-associated DPLD accompanied by multiple cystic lesions, multiple lymphadenopathy, and polyclonal hypergammaglobulinemia with elevated immunoglobulin G (IgG) and IgG4 levels. We recommend a close evaluation of MCD in cases of DPLD with hypergammaglobulinemia.
- Castleman disease (CD) is an uncommon lymphoproliferative disorder first described by Benjamin Castleman in 1956 [1]. Clinically, CD has three distinctive subtypes; unicentric CD (UCD), human herpesvirus-8 (HHV-8)-associated multicentric CD (MCD), and idiopathic MCD (iMCD) [2]. MCD is characterized by generalized weakness, anemia, generalized lymphadenopathy, polyclonal hypergammaglobulinemia, high erythrocyte sedimentation rate (ESR), and increased C-reactive protein (CRP) levels as a consequence of elevated interleukin-6 (IL-6) levels [3]. Immunoglobulin G4-related disease (IgG4-RD) typically manifests as tumor-like enlargement of exocrine glands or extranodal tissues with an elevated serum IgG4 level. IgG4-RD occasionally involves lymph nodes. It may mimic MCD histologically [4]. Langerhans cell histiocytosis (LCH) is related to the aberrant proliferation of the mononuclear phagocyte system and Langerhans cell infiltration. Diffuse parenchymal lung disease (DPLD) with multiple cystic lesions is a characteristic finding of pulmonary LCH. Although MCD is rarely associated with DPLD, it can be considered in patients presenting with systemic inflammatory manifestations [5]. The authors encountered a case of iMCD presenting as DPLD with multiple cystic lesions accompanied by multiple lymphadenopathy and polyclonal hypergammaglobulinemia with elevated IgG and IgG4 levels. Thus, there is a need to differentiate MCD from IgG4-RD, LCH, and other DPLDs.
Introduction
- Ethical statements: This study was approved by the Institutional Review Board (IRB) of CHA Gumi Medical Center, CHA University (IRB No: GM 21-07), and written informed consent from the patient was waived by the IRB.
- A 32-year-old previously healthy man was referred to our hospital because of anemia and elevated ESR and CRP levels without any subjective symptoms. Without any laboratory abnormalities except for high ESR/CRP levels and anemia, upper and lower endoscopies and bone marrow studies were conducted. There were no abnormal findings. During an observational follow-up of symptomatic changes, the patient presented with insidious and progressive generalized weakness, light-headedness, and dyspnea on exertion (DOE) for more than 1 year. Routine medical examination at a local medical center revealed newly developed multiple reticular infiltrations and cystic lesions in both lung fields, as well as anemia and high levels of ESR/CRP. The patient was transferred to our hospital for further evaluation of DPLD. On admission, the patient appeared chronically ill and slightly lethargic and complained of generalized weakness and loss of appetite without a specific past medical history other than chronic anemia.
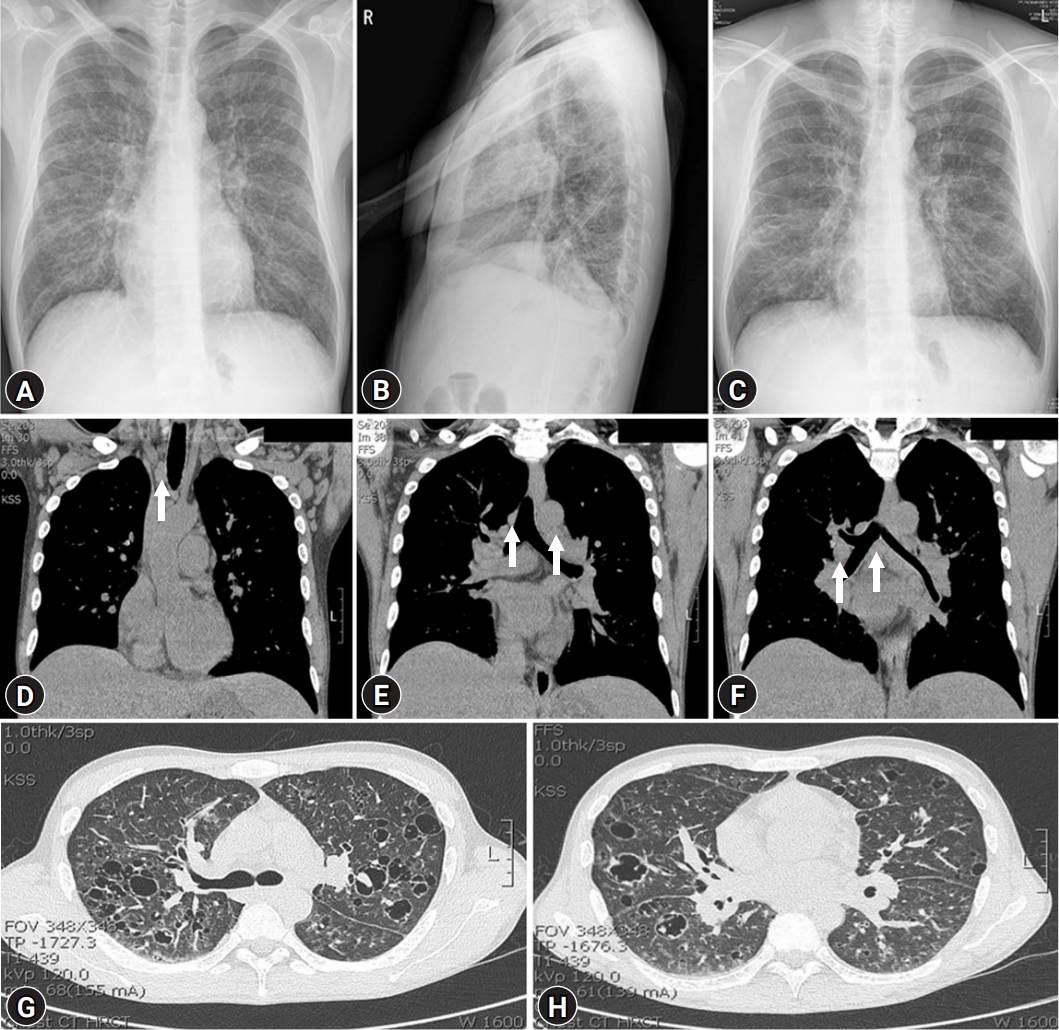
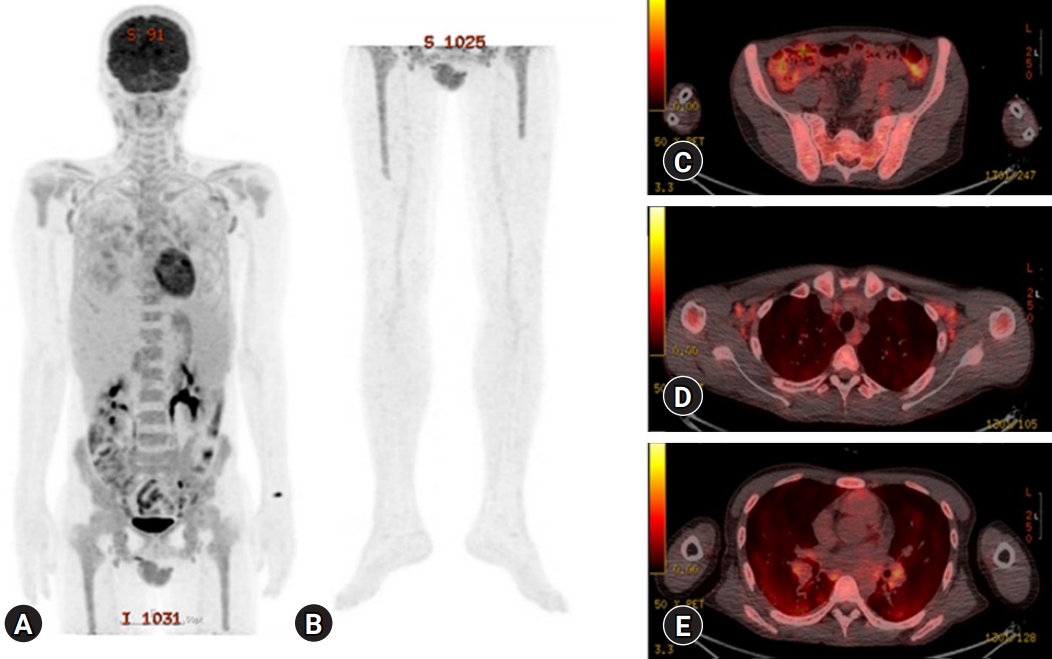
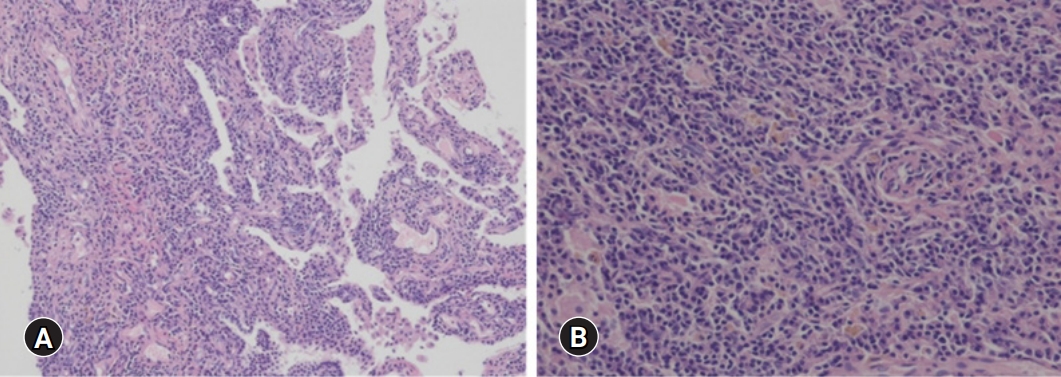
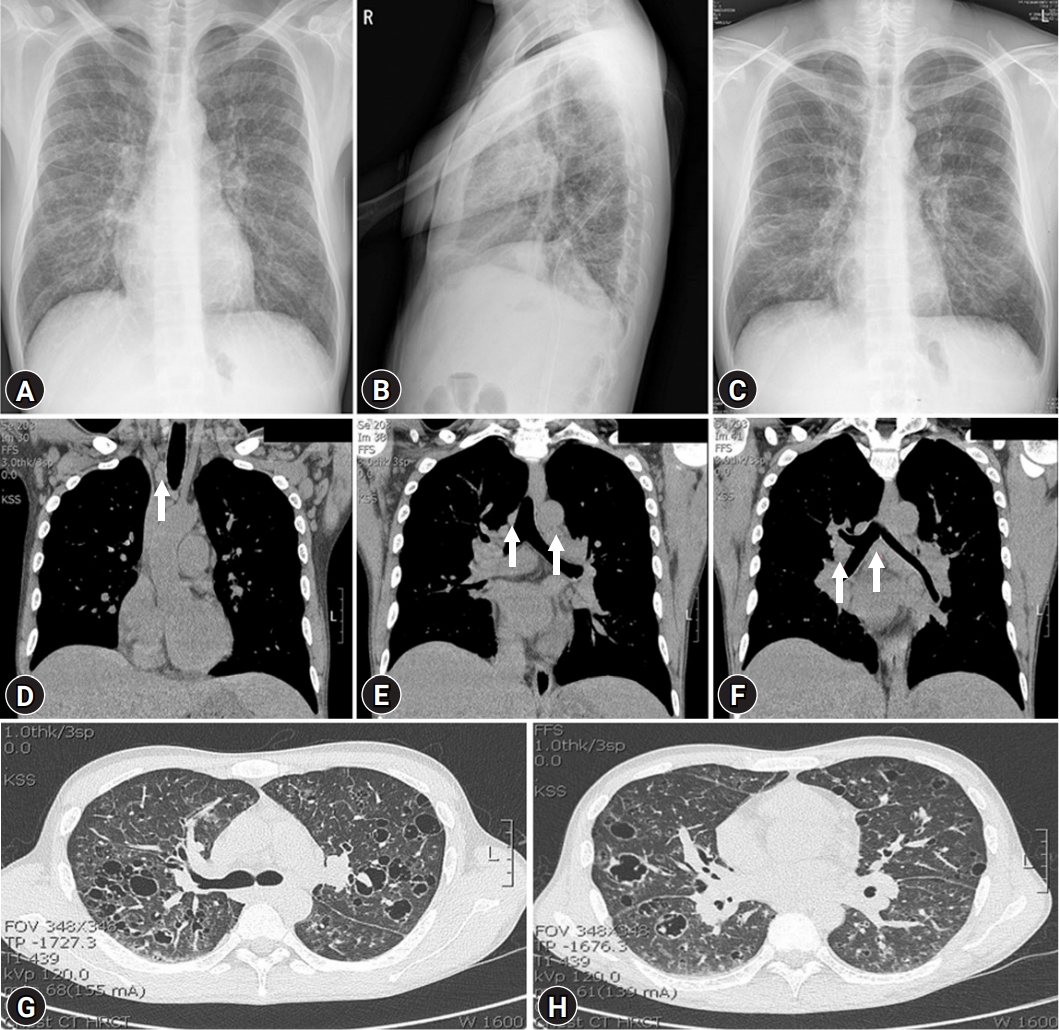
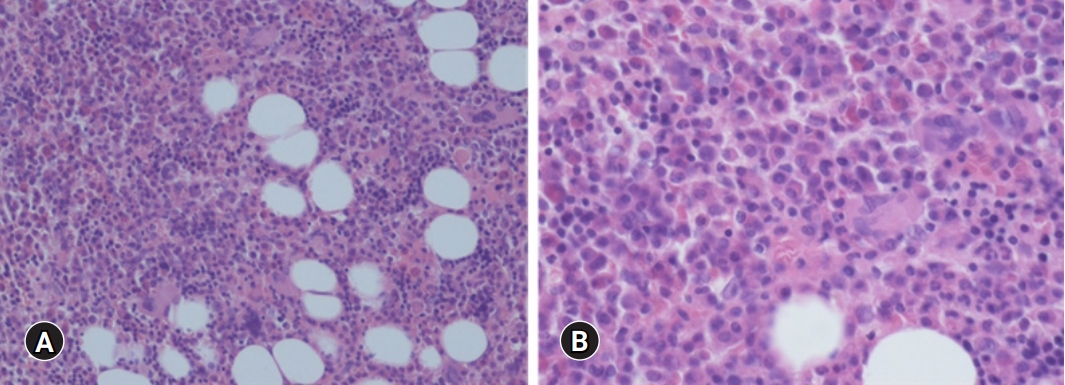
- His blood pressure was 120/80 mmHg, heart rate was 92 beats/min, respiratory rate was 20 breaths/min, and body temperature was 37.3°C. Initial laboratory tests revealed a white blood cell (WBC) count of 6,770/μL, hemoglobin (Hb) of 6.5 g/dL, platelet count of 584,000/μL, ESR of 40 mm/hr, CRP of 16.5 mg/dL, serum total protein (TP) of 12.09 g/dL, serum albumin (Alb) of 1.79 g/dL, Alb/globulin (A/G ratio) of 0.2, IgG8 of 190 mg/dL (range, 700–1,600 mg/dL), IgG4 subclass of 2,098 mg/L (range, 39.2–864 mg/L), IL-6 of 117.1 pg/mL (≤8 pg/mL), and HHV-8 polymerase chain reaction negative. Protein electrophoresis results showed polyclonal gammopathy without monoclonal paraproteinemia, consistent with IgG-predominant hypergammaglobulinemia and anemia of chronic inflammation. Chest X-rays showed dense infiltrations in both lung fields (Fig. 1A–1C). Chest computed tomography (CT) scan revealed multifocal ground-glass opacity with the progression of multiple variable-sized cystic lesions as compared with that of a local medical center. There were multiple enlarged lymph nodes in the neck (I to V levels), axillae, upper and lower paratracheal, hilar, subaortic, and subcarinal areas (Fig. 1D-1H). These lesions were radiologically diagnosed as pulmonary LCH. The patient underwent positron emission tomography-CT (PET-CT) for the evaluation of lymphoproliferative diseases, lymphoma, and other malignancies. The 18F-fluorodeoxyglucose uptake was diffusely enhanced in both lung parenchyma, bone marrow of the pelvis to femurs, and multilevel lymph nodes throughout the body on a PET-CT scan (Fig. 2). To evaluate polyclonal hypergammaglobulinemia and anemia, bone marrow aspiration and biopsy were performed. Bone marrow studies revealed that hypercellular marrow particles (80% cellularity), myeloid, and erythroid cells were normally mature and megakaryocytes were mildly increased in number. However, plasma cells were markedly proliferated up to 11% based on the absolute neutrophil count (Fig. 3).
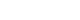
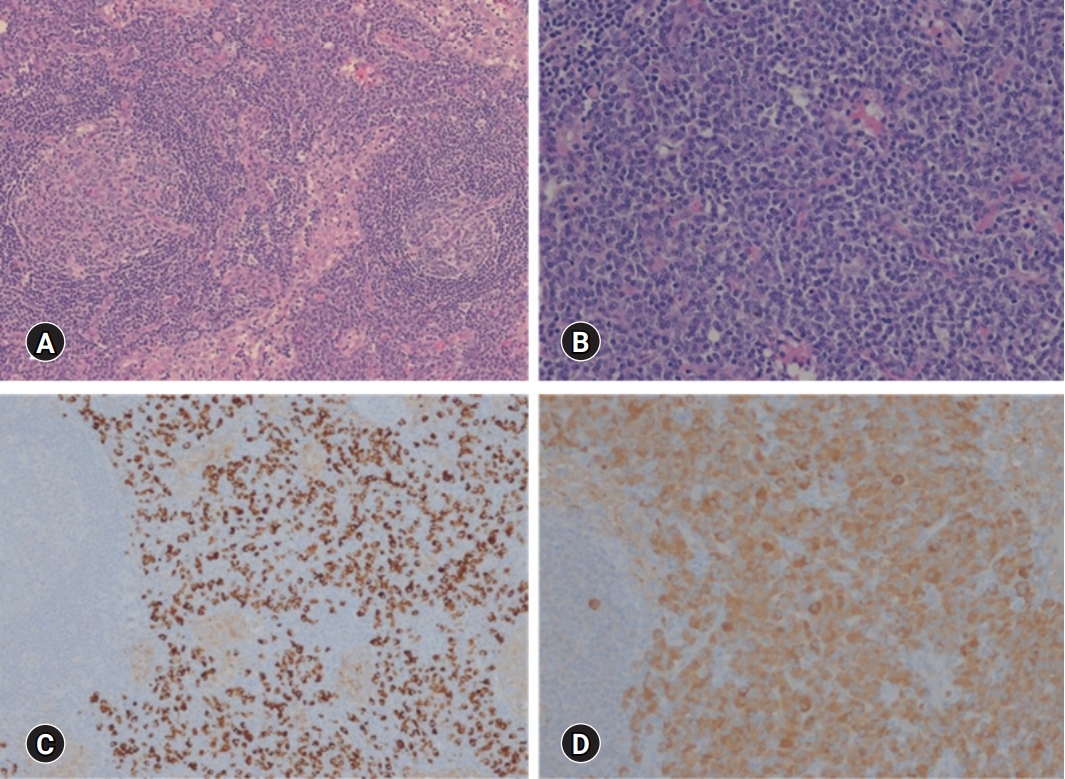
- An excisional biopsy was performed targeting the left external inguinal lymph nodes. The biopsy showed marked interfollicular plasmacytosis and regressed germinal centers (Fig. 4A, 4B) with IgG4 positivity of more than 100/high power field (HPF) and 70% on immunohistochemical staining (Fig. 4C, 4D). Finally, the patient underwent video-assisted thoracic surgery with wedge lung resections of the right middle and lower lobes. Histopathological findings showed dense plasmacytic and histiocytic infiltrations in the interstitium with multifocal lymphoid aggregates (Fig. 5). However, there was no evidence for the presence of Langerhans cells, eosinophils, neutrophil-forming giant cells, or abscesses. Immunohistochemical staining revealed that IgG and IgG4 positive cells were more than 400/HPF and 100/HPF, respectively. They were negative for HHV-8 and S-100 proteins.
- In addition to the above examinations, the patient tested negative for HHV-8, Epstein-Barr virus, cytomegalovirus, and toxoplasmosis. He was negative for autoimmune/autoinflammatory disorders such as systemic lupus erythematosus, rheumatoid arthritis, and adult-onset still disease. The laboratory results showed fluorescent anti-nuclear antibody titer of 1:40, dsDNA IgG of 13 IU/mL (<15 IU/mL), anti-ribonuclear protein antibody (Ab) of 0.53 U/mL (<10 U/mL), and anti-Smith Ab of 0.3 U/mL (<10 U/mL). The patient was also negative for polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) syndrome. Based on germinal center regression and plasmacytosis with histopathologic features of enlarged lymph nodes at ≥2 different sites, elevated ESR/CRP, anemia, thrombocytosis, hypoalbuminemia, and polyclonal hypergammaglobulinemia on laboratory tests accompanied by constitutional symptoms, hepatosplenomegaly, and lymphocytic interstitial pneumonitis, plasma cell histopathologic subtype of HHV-8-negative iMCD-complicated DPLD was diagnosed in accordance with the International, evidence-based consensus diagnostic criteria for HHV-8-negative/iMCD [6] and the 2019 American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) Classification criteria for IgG4-RD [7]. High-dose prednisolone therapy (60 mg/day) was initiated. With high-dose glucocorticoid monotherapy, symptoms of generalized weakness and DOE showed improvement. However, responses were partial and limited in a dose-dependent manner after 4 weeks of treatment. Contrary to the improvement in symptoms, no radiological improvement was observed on chest radiography. Responses based on follow-up laboratory tests were very subtle, showing a WBC count of 14,080/μL, Hb of 10 g/dL, platelet count of 695,000/μL, ESR of 120 mm/hr, CRP of 16.9 mg/dL, TP of 10.9 g/dL, Alb of 1.97, A/G ratio of 0.2, IgG of 6,698 mg/dL, and IgG4 subclass of 1,523 mg/dL. Without any significant improvement on glucocorticoid monotherapy, an IL-6 inhibitor, siltuximab (11 mg/kg, every 3 weeks), was administered while tapering prednisolone. On continuing siltuximab administration with low dose prednisolone (5 mg/day), laboratory tests resulted in Hb of 12.2 g/dL, CRP of 10.1 mg/dL, TP of 9.9 g/dL, Alb of 3.08 g/dL, and A/G ratio of 0.5. Although multiple variable-sized cystic lesions in both lungs were observed persistently, ill-defined patchy and nodular ground-glass attenuations and small well-defined nodules were decreased in both extent and size. On a follow-up chest CT, regression of enlarged lymph nodes was observed in bilateral neck, axillae, and mediastinum. After treatment for more than 30 months, the patient has maintained a favorable partial response to siltuximab without further progression or complications.
Case
- CD is a lymphoproliferative disorder characterized by generalized lymphadenopathy and systemic inflammatory manifestations. CD can be diagnosed by excluding CD-like lesions in the context of other diseases such as lymphoma, POEMS syndrome, primary lymph node plasmacytoma, follicular dendritic sarcoma, systemic lupus erythematosus, Sjögren syndrome, and IgG4-RD. Three distinct histological types are considered at diagnosis according to the features of the affected lymph nodes; hyaline-vascular type, plasma cell type, and mixed type [6,8]. Clinically, UCD is a localized disease. It usually exhibits hyaline vascular morphological changes with an excellent prognosis, whereas MCD is a systemic disease with polyclonal plasmacytic proliferation, probably due to an immunoregulatory deficit involving inflammatory signs such as lymphadenopathy, fever, splenomegaly, edema, weight loss, polyclonal lymphoproliferation, and life-threatening multiple organ dysfunction [6,9] due to IL-6 overproduction from activated plasma cells [10]. MCD is most commonly associated with HHV-8 or HIV. HHV-8 or the HIV-free state can be designated as iMCD.
- CD-associated DPLDs are rare and have not been well reported [5]. However, one study based on chest CT scans has suggested that CD-associated DPLD might be more prevalent and that a well-designed prospective study should be performed [5].
- In the present case, the patient visited the hospital several years ago for the evaluation of anemia and elevated ESR and CRP levels without subjective symptoms. However, during the observation period, generalized weakness and DOE developed over a 1-year period. No other infections, autoimmune/autoinflammatory disorders, hematologic diseases, or malignancies were found except for profound anemia, polyclonal hypergammaglobulinemia, and high ESR/CRP levels. A chest CT scan revealed ground-glass opacity with multiple cystic lesions consistent with LCH.
- Cystic lesions in both lungs and radiologic findings were consistent with pulmonary LCH. LCH is characterized by aberrant function and proliferation of cells of the mononuclear phagocyte system and infiltration of organs by Langerhans cells [11]. Typical pathologic findings of LCH are heterogeneous collections of Langerhans cells with eosinophils, neutrophils, small lymphocytes, and histiocytes that might form multinucleated giant cells. Occasionally, eosinophilic abscesses might be present, demonstrating central necrosis and positive S-100 protein, CD1a, and HLA-DR in immunohistochemical stains [11]. However, in the present case, lung biopsy showed no presence of Langerhans cells and negative staining for S-100 protein. Bone marrow aspiration and biopsy revealed hypercellular marrow (cellularity of 80%) with an increase in plasma cells (11.0%). There was no evidence of Langerhans cell disease on lymph node biopsy.
- An excisional biopsy of the lymph node showed interfollicular marked plasmacytosis, regressed germinal centers with IgG4 positivity of more than 70%, and 100/HPF on immunohistochemical staining. Wedge resections of the lung histologically demonstrated dense plasmacytic and histiocytic infiltration in the interstitium with multifocal lymphoid aggregates and negative immunohistochemical staining of HHV8 and S-100 protein. However, IgG and IgG4 positive cells were as many as 400/HPF and 100/HPF, respectively.
- IgG4-RD is pathologically characterized by diffuse lymphoplasmacytic infiltration, occasional eosinophilic infiltration, irregular fibrosis, and obliterative phlebitis with infiltration of IgG4-positive plasma cells into affected tissues, including the lungs [12]. Based on radiological, histological, and clinical features, the patient was evaluated for differential diagnoses of LCH, IgG4-RD, and iMCD. In accordance with the International, evidence-based consensus diagnostic criteria for HHV-8-negative/iMCD [6] and the 2019 ACR and EULAR Classification Criteria for IgG4-RD [7], clinical and laboratory findings were taken into consideration in association with histopathological features for the final diagnosis. Mochizuki et al. [13] reported overlapping features of IgG4-RD and MCD in a single patient treated with rituximab. Sato et al. [14] studied the clinical and pathological features of IgG4-related lymphadenopathy in comparison with hyper-IL-6 syndromes such as MCD, rheumatoid arthritis, and other immune-mediated conditions. Patients with hyper-IL-6 syndromes often fulfill the criteria for IgG4-RD, showing the same histological findings. As for the differential diagnosis, Sato et al. [14] have suggested that laboratory analyses are crucial and that hyper-IL-6 syndromes are characterized by high levels of serum gamma globulin and CRP, thrombocytosis, anemia, hypoalbuminemia, hypergammaglobulinemia, and hypocholesterolemia, which is not true for IgG4-RD. Our patient’s serum levels of IL-6, immunoglobulins, and CRP were consistently high along with thrombocytosis and anemia, suggesting MCD clinically.
- Based on histopathological and laboratory results along with clinical features, the patient was diagnosed with a plasma cell histopathologic subtype of HHV-8-negative iMCD-complicated DPLD. Medical therapy for MCD includes the use of glucocorticoids, monoclonal antibodies (mAb) against IL-6, chemotherapy, immune-modulators/suppressants such as thalidomide, immunoglobulin, rituximab, and hematopoietic cell transplantation [15]. With high-dose prednisolone, patient’s weakness, DOE, laboratory findings of anemia, high levels of ESR/CRP, and hypergammaglobulinemia started to improve. However, the effects were partial, dose-dependent, and limited, without radiological pulmonary improvements. Glucocorticoids are frequently used as systemic therapy for patients with iMCD. They can lead to mild symptomatic improvement during acute exacerbations of iMCD. However, glucocorticoid monotherapy was found to have no significant effect on the disease. Disease relapse was observed during tapering.
- According to the International, evidence-based consensus treatment guidelines for iMCD [16], siltuximab is recommended as initial therapy for patients with non-severe HHV-8 negative iMCD. Tocilizumab can be used if siltuximab is not available. Patients who do not respond to siltuximab or tocilizumab should be considered for rituximab-based therapy in combination with steroids. Patients with severe iMCD should be treated with siltuximab and high-dose steroids. If no clear response occurs within one week, combination chemotherapy should be considered. Patients with severe iMCD must have at least two of the following five criteria: (1) European Cooperative Oncology Group performance score of ≥2, (2) stage IV renal dysfunction (estimated glomerular filtration rate of <30 mL/min/1.73 m2), (3) creatinine of >3.0, (4) anasarca and/or ascites and/or pleural/ pericardial effusion, and (5) Hb of ≤8.0 g/dL pulmonary involvement/interstitial pneumonitis with dyspnea.
- The mAb targeting IL-6 (siltuximab) or the IL-6 receptor (tocilizumab, atlizumab) can be used in iMCD without POEMS syndrome. In a prospective randomized placebo-controlled trial investigating the efficacy of IL-6-neutralizing mAb siltuximab on iMCD [17], the median treatment duration for 19 patients was 5.1 years (range, 3.4–7.2 years), with 14 patients (74%) treated for more than 4 years. All iMCD patients in this extension study received siltuximab for a prolonged duration (up to 7 years) without evidence of cumulative toxicity or treatment discontinuation. They showed sustained disease control.
- In the aspect of prognosis, IgG4-RD is known to be typically responsive to steroid therapy [18]. In one study with 10 cases of IgG4-related lung disease, all patients except one were effectively treated with prednisolone alone, and the remaining patient responded to cyclosporine [19]. In contrast, the prognosis of MCD is generally poor. In a review series of MCD, the 2-year survival was 88% (95% confidence interval, 81%–95%) for a total of 114 patients with a median follow-up period of 29 months [19]. The most common causes of death in MCD are organ failure, sepsis, malignancy, and disease progression [20].
- In the present case, the patient tolerated combination therapy of siltuximab (11 mg/kg, every 3 weeks) and prednisolone (5 mg/day), showing no disease progression or any complication for 4 years.
- We report a case of iMCD presenting with atypical lung manifestations mimicking LCH and IgG4-RD with IgG/IgG4 dominant hypergammaglobulinemia. Our results suggest that further evaluation of MCD should be considered in the case of DPLD with hypergammaglobulinemia, although with low probability.
Discussion
-
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
-
Funding
None.
-
Author contributions
Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Visualization, Investigation, Resources, Software, Supervision, Validation: HJK, YHH; Writing - original draft: HJK, YHH; Writing - review & editing: HJK, YHH.
Notes


- 1. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956;9:822–30.ArticlePubMed
- 2. Fajgenbaum DC, Shilling D. Castleman disease pathogenesis. Hematol Oncol Clin North Am 2018;32:11–21.ArticlePubMed
- 3. Shahidi H, Myers JL, Kvale PA. Castleman’s disease. Mayo Clin Proc 1995;70:969–77.ArticlePubMed
- 4. Takenaka K, Takada K, Kobayashi D, Moriguchi M, Harigai M, Miyasaka N. A case of IgG4-related disease with features of Mikulicz’s disease, and retroperitoneal fibrosis and lymphadenopathy mimicking Castleman’s disease. Mod Rheumatol 2011;21:410–4.ArticlePubMed
- 5. Huang H, Feng R, Li J, Song X, Li S, Xu K, et al. Castleman disease-associated diffuse parenchymal lung disease: A STROBE-compliant retrospective observational analysis of 22 cases in a tertiary Chinese hospital. Medicine (Baltimore) 2017;96:e8173.ArticlePubMedPMC
- 6. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017;129:1646–57.ArticlePubMedPMC
- 7. Wallace ZS, Naden RP, Chari S, Choi H, Della-Torre E, Dicaire JF, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol 2020;72:7–19.ArticlePubMed
- 8. Oksenhendler E, Boutboul D, Fajgenbaum D, Mirouse A, Fieschi C, Malphettes M, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol 2018;180:206–16.ArticlePubMed
- 9. Bowne WB, Lewis JJ, Filippa DA, Niesvizky R, Brooks AD, Burt ME, et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer 1999;85:706–17.ArticlePubMed
- 10. Palestro G, Turrini F, Pagano M, Chiusa L. Castleman’s disease. Adv Clin Path 1999;3:11–22.PubMed
- 11. Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med 2000;342:1969–78.ArticlePubMed
- 12. Inoue D, Zen Y, Abo H, Gabata T, Demachi H, Kobayashi T, et al. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology 2009;251:260–70.ArticlePubMed
- 13. Mochizuki H, Kato M, Higuchi T, Koyamada R, Arai S, Okada S, et al. Overlap of IgG4-related disease and multicentric Castleman’s disease in a patient with skin lesions. Intern Med 2017;56:1095–9.ArticlePubMedPMC
- 14. Sato Y, Kojima M, Takata K, Morito T, Asaoku H, Takeuchi T, et al. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol 2009;22:589–99.ArticlePubMed
- 15. van Rhee F, Voorhees P, Dispenzieri A, Fosså A, Srkalovic G, Ide M, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018;132:2115–24.ArticlePubMedPMC
- 16. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003;38:982–4.ArticlePubMed
- 17. Yamashita K, Haga H, Kobashi Y, Miyagawa-Hayashino A, Yoshizawa A, Manabe T. Lung involvement in IgG4-related lymphoplasmacytic vasculitis and interstitial fibrosis: report of 3 cases and review of the literature. Am J Surg Pathol 2008;32:1620–6.ArticlePubMed
- 18. Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol 2016;3:e163–75.ArticlePubMed
- 19. van Rhee F, Greenway A, Stone K. Treatment of idiopathic Castleman disease. Hematol Oncol Clin North Am 2018;32:89–106.ArticlePubMed
- 20. van Rhee F, Casper C, Voorhees PM, Fayad LE, van de Velde H, Vermeulen J, et al. A phase 2, open-label, multicenter study of the long-term safety of siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget 2015;6:30408–19.ArticlePubMedPMC
 PubReader
PubReader ePub Link
ePub Link Cite
Cite